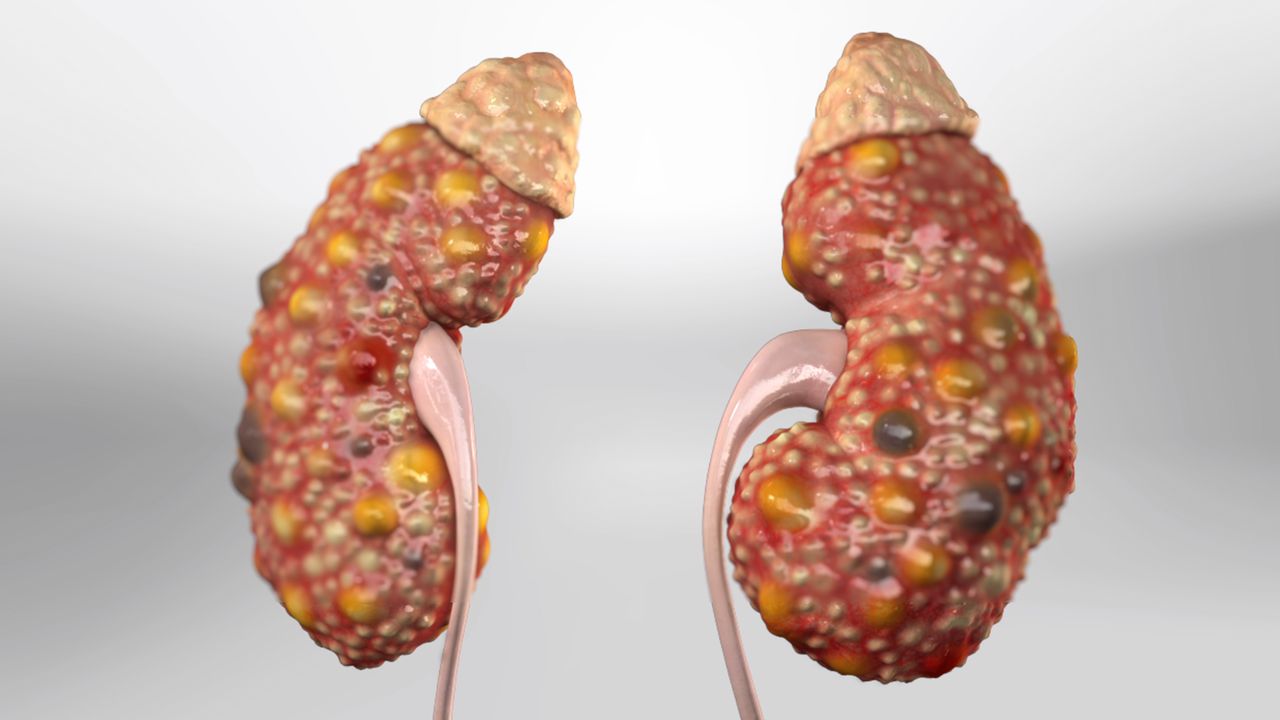
What is
polycystic kidney disease?
Polycystic kidney disease (PKD) is an inherited condition in which the kidneys gradually develop hundreds or even thousands of fluid-filled cysts. As the cysts grow, they replace functional kidney tissue. The kidneys enlarge — sometimes dramatically — and over years to decades, kidney function declines.
PKD is not the same as having a single kidney cyst, which is common and usually harmless. PKD is a systemic, inherited condition where cyst formation continues throughout life and affects both kidneys. It often affects other organs too — particularly the liver, occasionally the pancreas, and (importantly) the blood vessels of the brain.
The prognosis varies. Some people maintain reasonable kidney function into their 70s with appropriate management. Others progress to end-stage kidney disease in their 40s or 50s and need dialysis or transplantation. The variables that determine speed of progression — blood pressure control, lifestyle, hydration, and certain medications — are largely controllable.
Two genetic patterns —
very different timelines.
Autosomal Dominant PKD (ADPKD)
The far more common form. Symptoms usually appear in adulthood (30s–50s). A child of a parent with ADPKD has a 50% chance of inheriting it. Caused mainly by mutations in PKD1 or PKD2 genes.
Autosomal Recessive PKD (ARPKD)
Much rarer. Symptoms appear in infancy or childhood. Both parents must carry the gene (usually asymptomatic carriers). Often associated with liver disease as well. More aggressive disease course.
PKD1 vs PKD2 (within ADPKD)
PKD1 mutations cause faster progression — end-stage disease typically by 55. PKD2 mutations cause slower progression — end-stage disease typically by 70+. Knowing which gene matters for prognosis.
Acquired Cystic Kidney Disease
Not PKD, but worth distinguishing — patients on long-term dialysis can develop multiple kidney cysts that resemble PKD. Different cause, different management.
How PKD
typically appears.
In ADPKD, symptoms usually don't appear until adulthood. Many people are diagnosed in their 30s or 40s — sometimes after a parent has been diagnosed, sometimes after symptoms develop. Common presentations:
- Hypertension — often the earliest sign, sometimes in the 20s. About 50% of ADPKD patients have hypertension before any kidney function decline.
- Flank pain or fullness — from enlarging kidneys
- Blood in urine — from a ruptured cyst, occasionally from a stone
- Kidney stones — more common in PKD than in the general population
- UTIs — sometimes infections of individual cysts (particularly hard to treat)
- Enlarged abdomen — palpable kidneys in advanced disease
- Headaches — sometimes related to hypertension, occasionally a warning of intracranial aneurysm
- Liver cysts — common in ADPKD, usually asymptomatic but occasionally cause discomfort
- Family history — many patients are referred because a relative was diagnosed
How PKD is
identified.
The diagnosis of ADPKD is usually straightforward — imaging is the key. Genetic testing is occasionally needed but rarely the first step.
- Ultrasound — usually the first investigation. In a patient with family history, age-specific criteria apply: 3+ cysts (unilateral or bilateral) by age 30–39, 2+ in each kidney by 40–59, 4+ in each kidney by 60+.
- CT or MRI — more sensitive than ultrasound. Used when ultrasound is inconclusive, when measuring total kidney volume (an important prognostic marker), or when planning surgery.
- Blood tests — creatinine, eGFR, electrolytes, full blood count. Establishes baseline kidney function. Repeated at intervals to track progression.
- Urine analysis — protein, blood, infection markers.
- Genetic testing — used selectively. Helpful when imaging is equivocal, for prognosis (PKD1 vs PKD2), for family planning, or before kidney donation by a relative.
- Brain MRI/MRA — to screen for intracranial aneurysms in selected patients (family history of aneurysm, hypertension, planned major surgery).
By the end of the diagnostic workup, we usually know: the genetic type, the current kidney function, the size of the kidneys, the blood pressure status, and the presence or absence of complications. This determines the management plan.
When a family member
is diagnosed.
One of the most important aspects of PKD care extends beyond the patient. Each first-degree relative of someone with ADPKD has a 50% chance of being affected. Identifying them early matters — because the most effective intervention (blood pressure control from early adulthood) only works if started early.
Family screening protocol:
- Parents and siblings — should be offered ultrasound screening, even if asymptomatic. The diagnosis can be made by imaging alone in most cases.
- Children — usually screened by ultrasound from late teens. Earlier screening (in young children) is sometimes done if hypertension or symptoms appear.
- Adult children of an affected parent — best screened in their 20s. A clear scan after age 30 makes ADPKD very unlikely.
- Living kidney donor candidates — when a relative is being considered as a kidney donor for an ADPKD patient, very thorough screening (including genetic testing if needed) is essential.
Family screening is a conversation, not just a test. There are implications for insurance, for family planning, for psychological wellbeing. We discuss these openly, and where genetic testing is being considered, we coordinate with a genetic counsellor.
Slowing progression —
what works.
Blood Pressure Control
Aggressive BP control (target below 130/80, sometimes lower) is the most important intervention. ACE inhibitors or ARBs are first-line. Started early, this alone can add decades to kidney lifespan.
Adequate Hydration
2.5–3 litres of water daily suppresses vasopressin and may slow cyst growth. Particularly important in Jaipur summers when dehydration is easy.
Low Salt, Moderate Protein
Sodium under 5g daily helps BP and reduces cyst-fluid accumulation. Moderate protein intake (not low, not high) protects kidney function.
Tolvaptan
A vasopressin receptor antagonist that slows cyst growth in rapidly progressing ADPKD. Used in selected patients — requires monitoring of liver function. Coordinated with nephrology.
NSAIDs and Nephrotoxins
Avoid long-term NSAID use, contrast agents where possible, herbal medications of uncertain composition. Each can accelerate kidney damage.
Multi-disciplinary Care
Urology, nephrology, cardiology, and sometimes hepatology coordination. Particularly important as disease progresses.
PKD-specific
problems we manage.
Some complications of PKD need specific urological attention:
- Cyst infection — different from a normal UTI. Antibiotics must penetrate cyst walls (fluoroquinolones, trimethoprim, ciprofloxacin). Sometimes needs CT-guided cyst drainage.
- Cyst haemorrhage — sudden bleeding into a cyst causes severe flank pain. Usually self-limiting with rest, pain control, and observation. Sometimes needs intervention.
- Kidney stones — more common in PKD. Treatment is similar to standard stone disease but requires careful pre-op imaging because of the altered kidney anatomy.
- Massive kidney enlargement — when kidneys become so large they cause abdominal discomfort, feeding difficulty, or compromise other organs. Rarely needs surgical reduction or nephrectomy.
- Pain management — chronic flank pain is common. Approached through positioning, physiotherapy, careful medication choice (avoiding NSAIDs), occasionally cyst aspiration or denervation.
- Liver cysts — common but usually asymptomatic. Massive symptomatic liver cysts may need drainage or, rarely, partial liver resection.
- Intracranial aneurysm — about 8–10% of ADPKD patients. Screening MRI is offered in selected patients. Detected aneurysms may need neurosurgical management.
- End-stage kidney disease — when eGFR drops below 15–20, planning begins for dialysis or transplantation.
Continuity, coordination,
long-term care.
Long-term relationship
PKD is a lifelong condition. We build the long-term clinical relationship needed for surveillance, family screening, and complication management.
Urology + nephrology coordination
PKD needs both specialties. We handle urological complications (stones, infections, cysts) and coordinate with nephrology partners for medical management and dialysis planning.
Family screening
Each affected patient triggers screening conversations for parents, siblings, and adult children. We organise and run the family screening process.
Transplant pathway coordination
For patients approaching end-stage disease, we coordinate with transplant centres for evaluation, living-donor workup, and listing.
Pricing for PKD
care & surveillance.
| Service | Starting from |
|---|---|
| PKD consultation | ₹ [____] |
| Ultrasound KUB (family screening or follow-up) | from ₹ [____] |
| MRI kidneys (volume measurement) | from ₹ [____] |
| Genetic testing (when indicated) | from ₹ [____] |
| Cyst aspiration (image-guided) | from ₹ [____] |
| Stone treatment (RIRS / PCNL) | from ₹ [____] |
| Annual surveillance package | from ₹ [____] |
| Family member screening visit | from ₹ [____] |
Hospitalisation covered. Surveillance often self-pay.
Hospitalisation for stone surgery, cyst complications, or other PKD-related admissions is covered by major insurers. OPD surveillance and family screening are usually self-pay. Genetic testing coverage varies by policy.
PKD —
your questions.
If your parent has ADPKD (the common form), you have a 50% chance of inheriting it. The best way to know is screening — usually an ultrasound from your 20s. A clear scan after age 30 makes ADPKD very unlikely.
No cure currently exists, but progression can be dramatically slowed. Optimal blood pressure control, adequate hydration, dietary measures, and (in selected patients) tolvaptan can add years or decades to kidney lifespan. When eventually needed, kidney transplantation is highly successful in PKD patients.
Some PKD patients reach end-stage disease (eGFR below 15) and need renal replacement therapy — typically in their 50s or 60s with PKD1, later with PKD2. Many patients never reach this stage. Optimal management delays the timeline significantly.
Yes — many PKD patients have children. The 50% inheritance risk is a personal decision. Pre-implantation genetic diagnosis (PGD) with IVF is an option for families wanting to avoid transmission. Genetic counselling helps with this decision.
The biggest impact comes from blood pressure control (medication + low salt). Next: hydration (2.5–3 litres daily, more in Jaipur summers). Then: avoiding NSAIDs and other nephrotoxins. Moderate protein intake. Weight management. Regular exercise. The unglamorous basics, done consistently.
Acute pain from a cyst can be from bleeding, infection, or rupture. Comes to clinic — we'll assess with ultrasound or CT and treat appropriately. Most cyst pain settles with rest and pain control. Persistent severe pain may need image-guided drainage.
Routine brain MRI is not recommended for all ADPKD patients, but is offered in specific situations — family history of brain aneurysm or haemorrhage, uncontrolled hypertension, before major surgery, or new-onset severe headaches. We discuss individual risk.
Hospital admissions for PKD-related complications are covered. Routine OPD surveillance is often self-pay. Tolvaptan and other PKD-specific medications may not be reimbursed by all policies — we tell you upfront.